Really proud to report that the first of our bona fide real-time phylogenomics papers is now out in Scientific Reports!
In the paper we managed to show a number of things that are potentially really exciting, and I’ll get to them in a minute. First though, this is the first paper I’ve published where I got to drive every part: from conceiving the idea (with Alex) to getting funding, planning and carrying out the fieldwork/field-sequencing (with Alex and Dion), sequencing (all by Andrew) and analysis and writing (everyone). This was incredibly satisfying as normally a lot of my time is spent analysing downstream data. I feel like a proper grown-up scientist now. More please!
Firstly, what did we actually do? Pretty simple really:
- Over a week (May 2016) in Snowdonia National Park, Wales,
- We collected the flower Arabidopsis thaliana and congeneric A. lyrata,
- Extracted their DNA and prepared sequencing libraries for MinION sequencing, in a tent with no mains power or running water,
- Sequenced both species using Oxford Nanopore MinION, and
- Analysed them in real-time with BLAST databases held locally, building trees with a handful of genes.
Later on back in the lab we repeated the sequencing (but not extractions) with Illumina MiSeq, so we could compare the platforms, and also developed a few more sophisticated bioinformatics analyses. To be honest, most of the pipelines we ran could have run in real-time (and now do) but at the time of the main fieldwork we just didn’t expect it would work as well as it did!
Result 1: Genomic DNA sequencing with MinION is fairly easy, even in the middle of nowhere.
Seriously, depending on how much patience and practical skill you have, this is either easy, or really easy. We used the Oxford Nanopore 1D Rapid sequencing kit (disclaimer: actually a prototype ONT provided; though the COTS one is much better now) for sequencing, and extracted using the Qiagen DNEasy Plant Miniprep kit, modified with a longer initial incubation and double-concentrated cleaning step, but essentially unchanged. The MinION itself, as is well documented, runs off USB into a laptop.
Hardware-wise you’ll need:
- Two waterbaths (or in our case, two polyboxes, a gas kettle, and some thermometers)
- A centrifuge
- A generator to run said devices
- Some poly boxes with -20ºC and ice for reagents
… that’s it. If you’re looking at this list and thinking ‘I could get all that together by next weekend, maybe we should go on a sequencing trip’ well, that’s the idea 🙂
There’s a lot of refinements possible. A portable freezer will make life easier, as will a dedicated 12v supply for USB power and a portable DNA quantification tool like a Quantus. Plus, all of the above don’t really like rain so a tent and/or van (as with Nick Loman and Josh Quick’s Zika trip last year) will help out a lot. But to get started, that’s it.
Result 2: Long MinION reads are really good at species ID – even better than Illumina in key respects.
The core goal of this project was to work out “can field-based genomic WGS sequencing identify closely-related species?”. So we deliberately picked two species from the same genus with publicly-available reference genome sequences (A. thaliana & A. lyrata). The ID process would be simple. For each of the four datasets (two MinION runs, one from each species, and two MiSeq 2x300bp paired-end runs, one for each species), we’d simply:
- Trim adapters from each read
- Match each read to the A. thaliana genome using the best hit with BLASTN
- Match each read to the A. lyrata genome, using the same method
- Compare the hit scores for each reference genome
- Un-blind the read (reveal which of the two species it actually came from)
- Score the read as a true or false positive or negative, depending on the result.
Clearly many reads finding a BLAST alignment for one species will also find a significant alignment to the other species, since these are separated by only a couple of MYr (and are pretty similar phenotypically). So if both hits are ‘significant’, how would we distinguish the best one? Intuitively it seems sensible that the longest match / most identities will be better, but what threshold length difference should we use? 1bp longer? 10bp? 100bp?
Happily the
package in R lets you investigate the performance of test statistics on known classifier sets. We used this to produce the plot below, which shows the effect of increasing threshold length difference on true-positive (TP), false-positive (FP) and accuracy rates for MinION (black) and MiSeq (red) reads: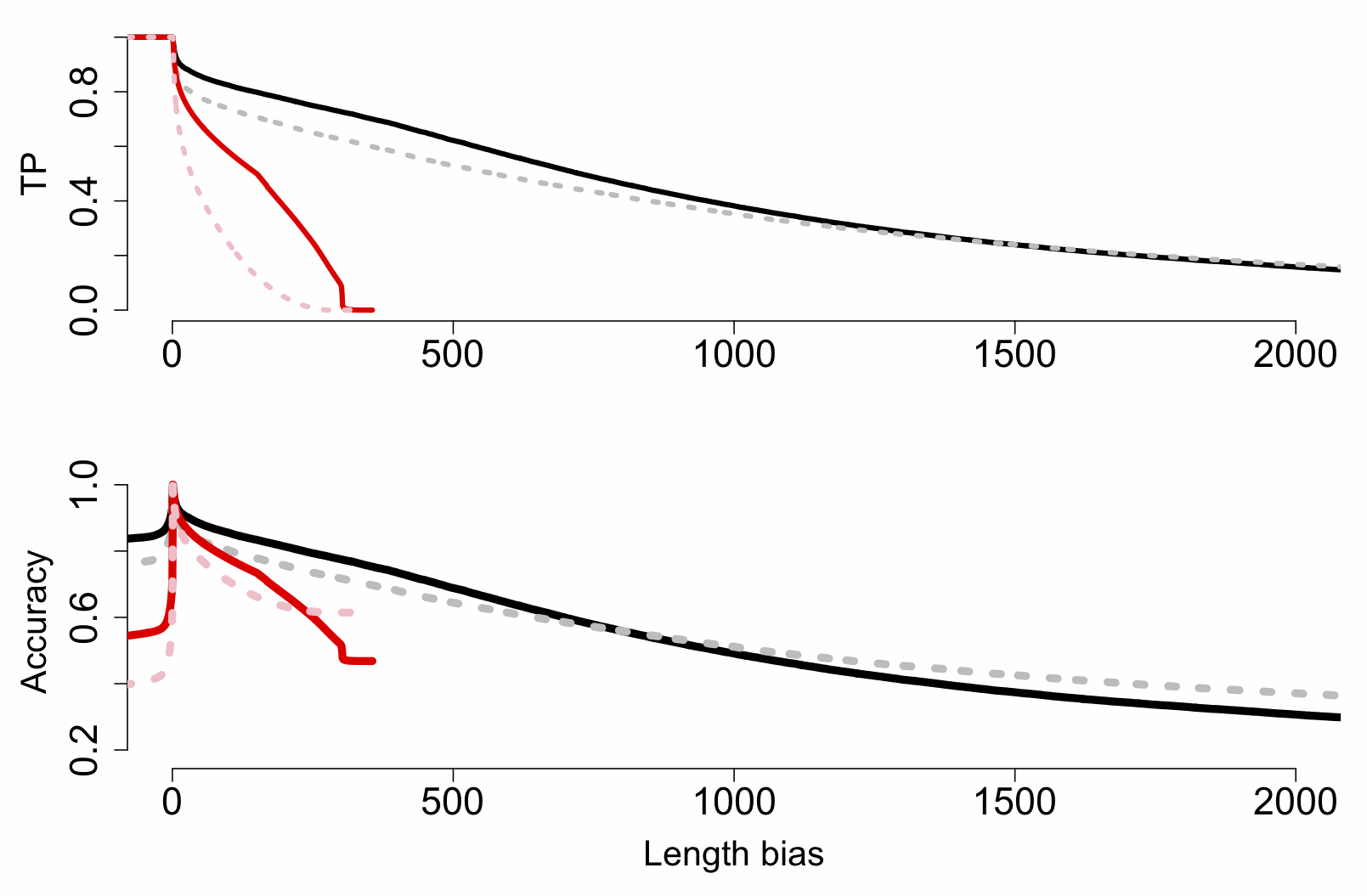
The really key thing here is that MinION reads are beating MiSeq ones at most length difference (bias) thresholds greater than ~5-10bp, right up to 300bp (the MiSeq inserts top out at this length, of course). This is important because while here we’re matching orthogonal ID cases (A. lyrata against A. thaliana, and vice versa), in a practical application we might have a third species without a reference but two possible matches, and while some loci will be closer to the first, others could match the second. So while a threshold of 1bp might technically be the best (TP rate of ~90% and close to 100% accuracy), we may want to raise the threshold to a much higher value (>50bp) and accept lower TP rates to get a better confidence.
Result 3: Species ID does not need complete genomes for reference databases, and even works with a handful of MinION reads.
One very obvious and sensible criticism of our early drafts of this was that the reference genomes we used to build BLASTN databases with are largely completely assembled. While there’s been some handwringing recently about the structural variation of these plant genomes at population level, most people accept that a high proportion of the informative sequences in these genomes are now well determined.
For most people, in most places, this will not be the case; for instance, there’s ~300,000-400,000 plant species described, but only ~180-250 public genomes. Most of those are of the fairly-low-coverage HTS WGS variety as well, so are pretty bittily assembled. Quite often these come from first-year baselining experiments in the ‘get some DNA and run it through the MiSeq then SOAP or Abyss’ mould, with N50 values in the low ‘000s.
So to test the effect of this, we artificially digested the reference genomes a few thousand times to simulate N50 values from about 100 (virtually unassembled) up to 10^6 (essentially complete), shown here in Fig 3a for N50 values from 10^0 up to 10^4, with accuracy scores calculated for a range of cutoff values:
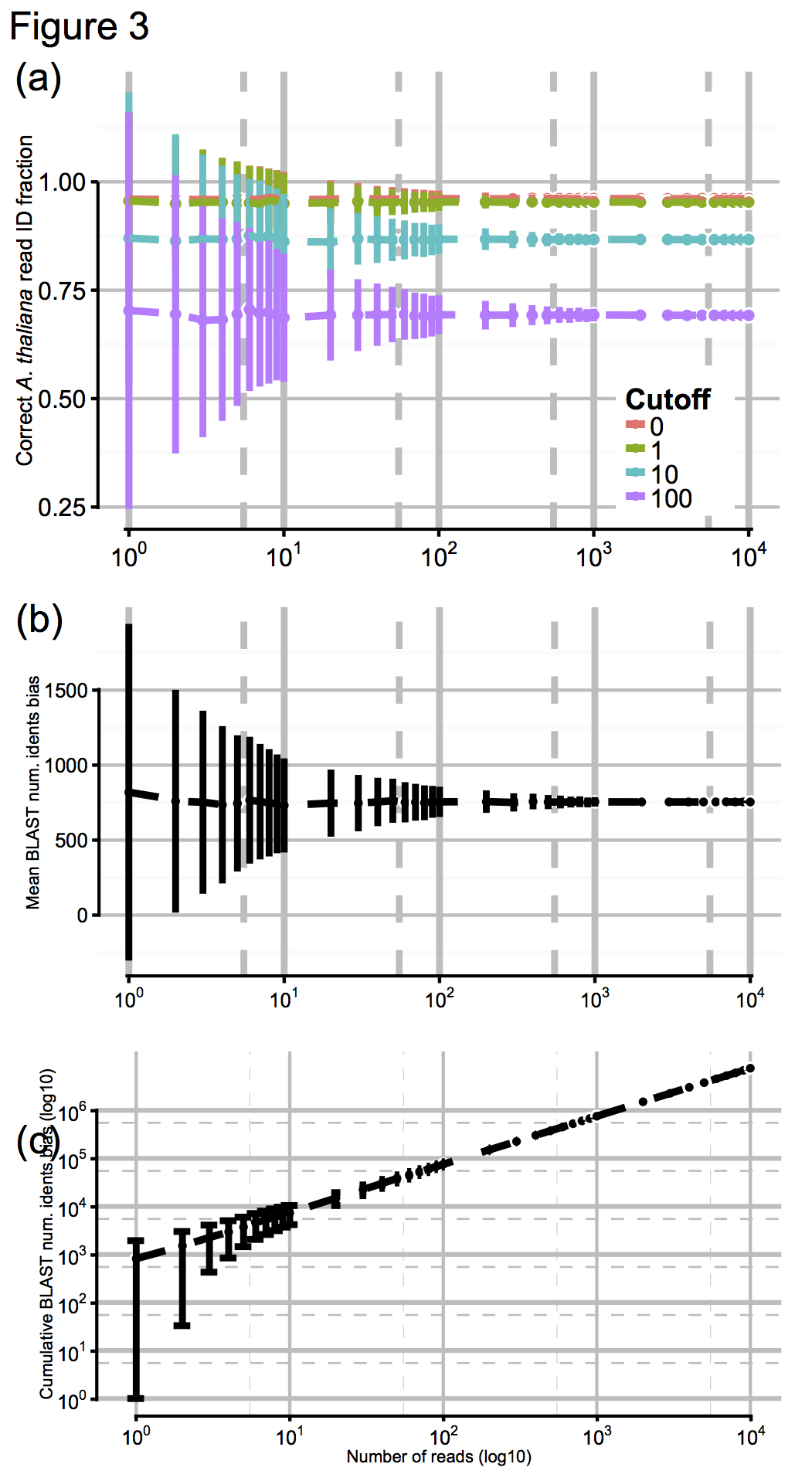
These results were pretty promising, so finally we asked ourselves: OK, we had tens of thousands of MinION reads to make our ID with, generated over a day or so: but how few reads we would need to have a stab at a correct ID? Again, we jacknifed our dataset to find out, shown above in Figs 3b and 3c. Promisingly, you can see that by about 10^2-10^3 reads (in practice, an hour or less) the confidence intervals on our ID score barely budge. So, after an hour of sequencing, you’re likely to get as good an answer as you can get. One. Hour…!!!
Result 4: Field-sequenced WGS MinION long-reads substantially improve downstream genomics with low-coverage HTS data.
When planning the fieldwork we hadn’t really known what we’d get, in terms of read length, quality, or yield: this was a prototype kit that not many people had played with yet, let alone taken into the field. But about the time we were writing this up we found out about various genome assemblers optimised (supposedly) for ONT reads, chiefly Canu and hybrid-SPAdes. We decided to give it a whirl.. the results are pretty amazing!
| Data |
MiSeq only |
MiSeq + MinION |
| Assembler |
Abyss |
hybridSPAdes |
| Illumina reads, 300bp paired-end |
8,033,488 |
8,033,488 |
| Illumina data (yield) |
2,418 Mbp |
2,418 Mbp |
| MinION reads, R7.3 + R9 kits,
N50 ~ 4,410bp |
– |
96,845 |
| MinION data (yield) |
– |
240 Mbp |
| Approx. coverage |
19.49x |
19.49x + 2.01x |
| Assembly key statistics: |
|
|
| # contigs |
24,999 |
10,644 |
| Longest contig |
90 Kbp |
414 Kbp |
| N50 contiguity |
7,853 bp |
48,730 bp |
| Fraction of reference genome (%) |
82 |
88 |
| Errors, per 100 kbp: #N’s |
1.7 |
5.4 |
| # mismatches |
518 |
588 |
| # indels |
120 |
130 |
| Largest alignment |
76,935 bp |
264,039 bp |
| CEGMA gene completeness estimate: |
|
|
| # genes |
219 of 248 |
245 of 248 |
| % genes |
88% |
99% |
Result 5: Individual MinION reads can be directly, individually annotated for coding loci with no assembly required.
By now everyone was getting a bit sick of me going on about MinION reads, but there was one final hunch I wanted to test: If reads are about the same length as nuclear coding loci (~5000-50,000bp), does that mean we can annotate individual reads to pull out coding sequences, and use them to build phylogenies? SNAP was a great tool for this, not least because it’s trained on A. thaliana gene models already.
I want to be absolutely clear here, as sometimes people seem to miss this: I’m not talking about assembling reads before annotation as usual. I’m not even talking about assembling them in real-time, then annotating. I mean, each time a read finishes basecalling, immediately try and annotate that single read, and only that single read, to try and get a coding sequence.
In other words, how quickly can we turn a tube of DNA, into a folder of sequence reads, into alignments of coding loci? The answer is, ‘bloody quickly’:
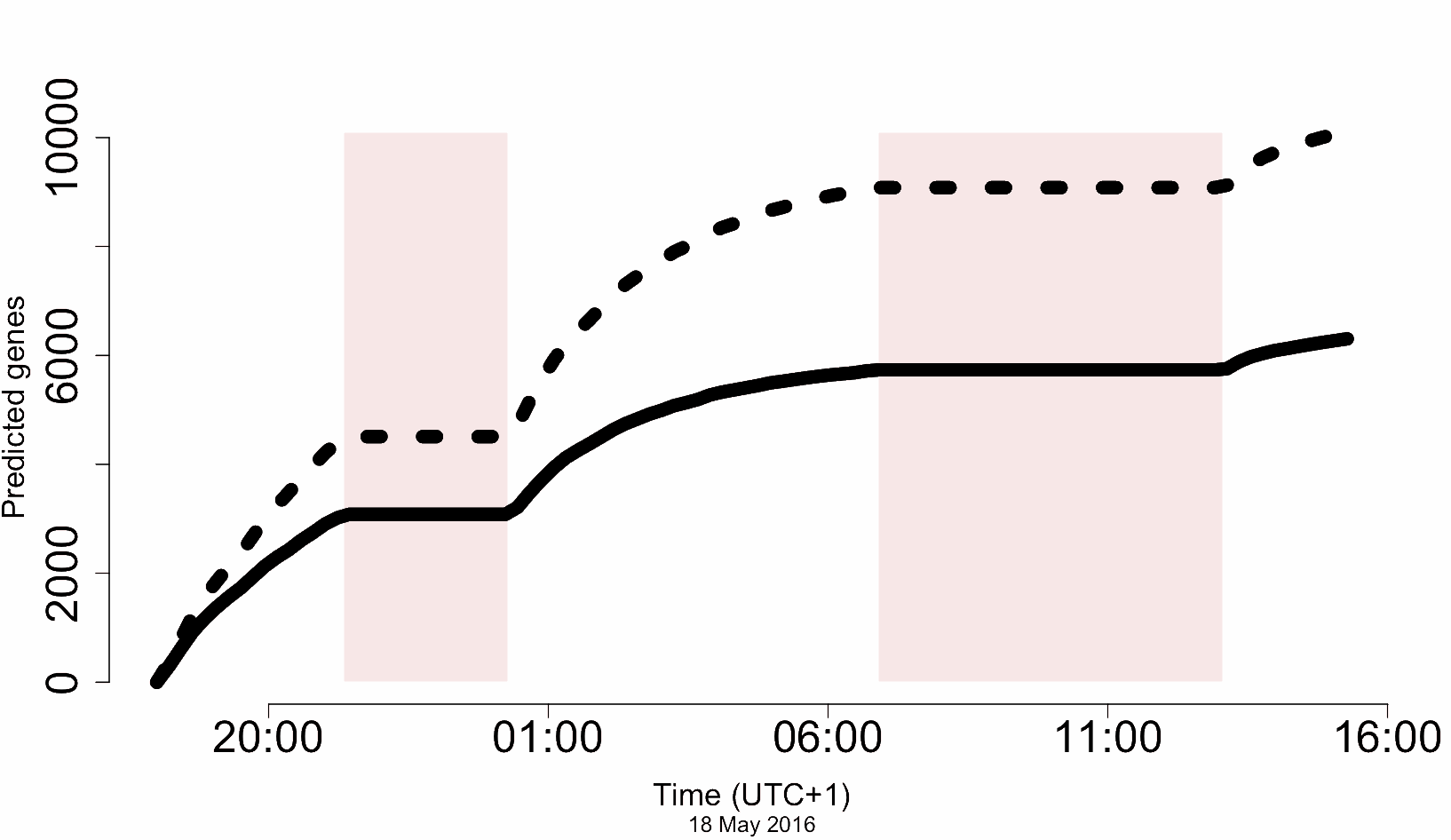
The dashed line shows ‘all gene models’. The solid line shows unique CDS. The axis is the number of predicted CDS and yes, those are thousands – recall that the total number of CDS for A. thaliana is only about 23,000. The axis is, well, actual sequencing time (!)*
Now, not all of these are complete genes, and error rate means distinguishing paralogs robustly in a real case (e.g. completely novel genome) would be tricky, but on the other hand, this was a completely unoptimised pipeline, really just hacked together over a couple of weeks. There’s a lot of scope to improve this…
*These are the read timestamps, but it wasn’t a live run. I actually ran the analysis back in the lab afterwards, as my code was too buggy on the fieldwork day and I just lost my shit. But the CPU demands aren’t high – I can and have run this live subsequently.
Result 6: Predicted coding loci from individual MinION reads can be aligned to orthologous sequences, and multilocus phylogeny inferred in real-time.
I build trees. I build trees. I build trees for a living. Did you seriously think, that having got as far as spitting out thousands of novel coding loci per day in field-based sequencing, I wasn’t going to try some real-time, field-based, multilocus phylogenomic inference?
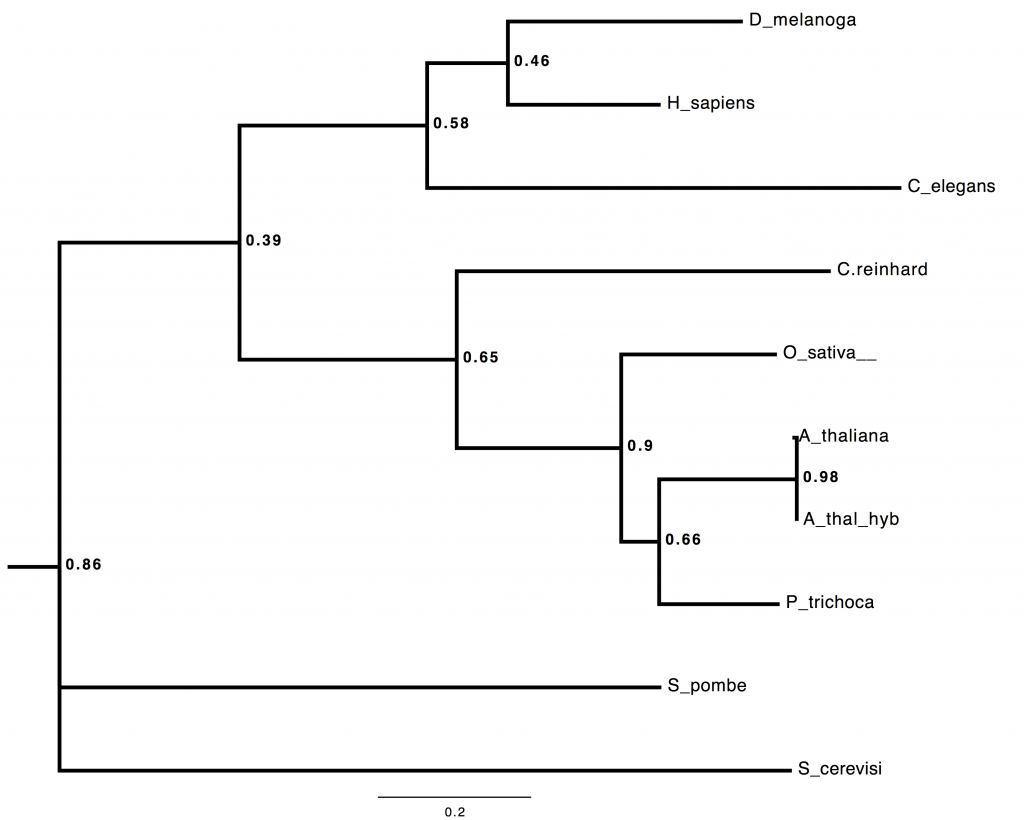
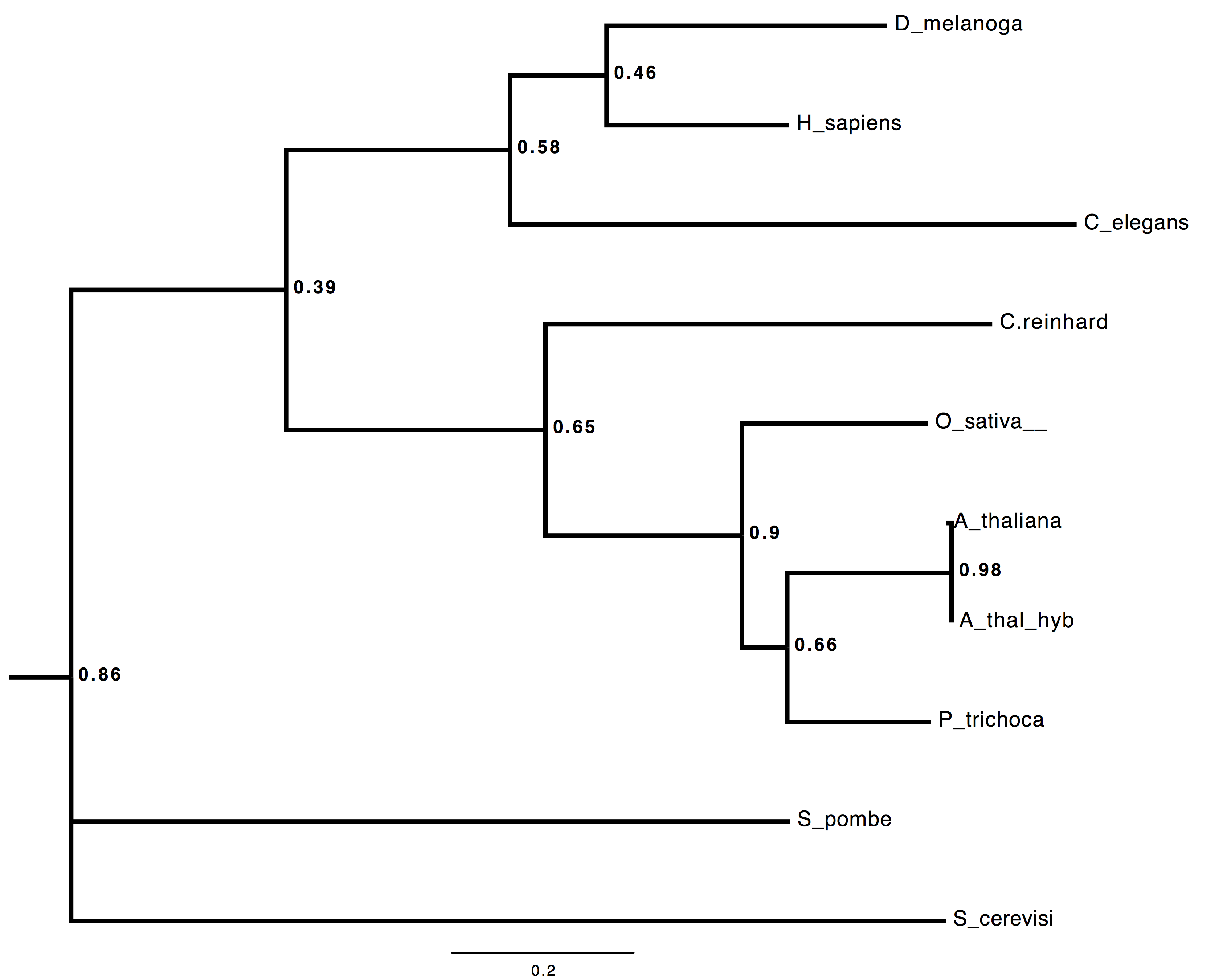
Here’s a *BEAST tree from 53 loci, all of the A. thaliana ones coming from directly-annotated, field-sequenced reads. Seriously 😉
Summary
As you’ve gathered by now, I’m enormously happy with this research. I think this paper is easily a bigger contribution to general science than our 2013 molecular convergence one because, if we’re honest, it’s an interesting but niche phenomenon, whereas literally everyone who uses or categorises any kind of biological material can benefit from this paper.
I’m indebted to my colleagues, Alex S. T. Papadopulos, Dion Devey, Andrew Helmstetter and Tim Wilkinson, and our funders, the Kew Foundation. We didn’t invent the MinION – that took hundreds of incredibly clever people at Oxford Nanopore years and millions of pounds of investment to do – but in this study we’ve managed to show all of the really transformative aspects of this technology working in the field, in real-time. There is no technical reason, at all why we shouldn’t all expect that within a decade, all of the analyses we currently run on DNA data in labs can run in the field, within minutes of collecting biological samples. And that really is something.