Talk presented at the #bench16 (benchmarking) symposium at KCL, London, Wed 20th April 2016. Funded by the SSI.
Slides (Slideshare – cc-by-nd)
[slideshare id=61147571&doc=joe-parker-benchmarking-bioinformatics-160420140811]

My last MinION post described our first experiments with this really cool new technology. I mentioned then that their standard library prep was fairly involved, and we heard that the manufacturers, Oxford Nanopore, were working on a faster, simpler library prep. We got in touch and managed to get an early prototype of this kit for developers*, so we thought we’d try it out. So our** experiments had three aims:
This is a lot of combinations to perform over a few days on one sequencing platform! Our experience from the last run gave us hope we could manage it all (we did, but with a lot of headaches over disk space – it turns out one drawback of being able to run multiple concurrent sequencing runs is hard-drive meltdown, unless you’re organised from the start – oops). In fact, to keep track of all the reads we added an ‘index’ function to the Poretools package. I really recommend you use this if you’re planning your own work.
We had eight samples to sequence, a 15-year-old dried fungarium specimen of Armillaria ostoye (likely to be poor quality DNA; extracted by Bryn Dentinger with a custom technique); some fresh Silene latifolia (Qiagen-extracted, which we’d used successfully with the previous, ‘2D’ library prep); and six arbitarily-selected plant samples, both monocots and dicots, extracted by boiling with Chelex beads (more about them at the end).

First we prepared normal ‘2D’ libraries from the Silene and Armillaria. These performed as expected from our December experiments, even the Armillaria giving decent numbers and lengths of reads (though not as many as we hoped, with some indication of worse Q-scores. We put this down to nicks in the fungarium DNA, and moved on to the 1D preps while the sequencer was still running.
The ‘1D’ in the rapid kit (vs ‘2D’ in the normal kit) refers to how many DNA strands are sequenced; in the 2D version, both forward and reverse-complement strands are sequenced. This is slower to prepare (extra adapters etc to link the two strands for sequencing) and also runs through the MinION more slowly (twice as much DNA, plus a hairpin moment) but is roughly twice as accurate, since each base is read twice. The 1D kit, on the other hand, results in single-stranded fragments, meaning we could expect lower accuracy traded off for higher speed. And the rapid kit really was fast – starting from purified extracted DNA, we added all the necessary adapters for sequencing in well under 15mins, ready to sequence.
The sequencing itself (for both the Armillaria and the Silene) went spectacularly well. Remember, this is unsheared genomic DNA, and imagine our surprise when we started to see 25, then 50, then 100, then 150kb reads come off the sequencer – many mapping straight away to reference genomes! It turns out that the size distribution of the 1D prep is much more long-tailed than the 2D/g-TUBE one. In fact, whereas the the 2D library looks like a normal-ish gamma, the 1D reads are more like an inverse exponential – lots of short stuff and then a very long tail with some mega-whopper reads in. Reads so long, in fact, that mapping them the same way as Illumina short-read data would be a bit bonkers…
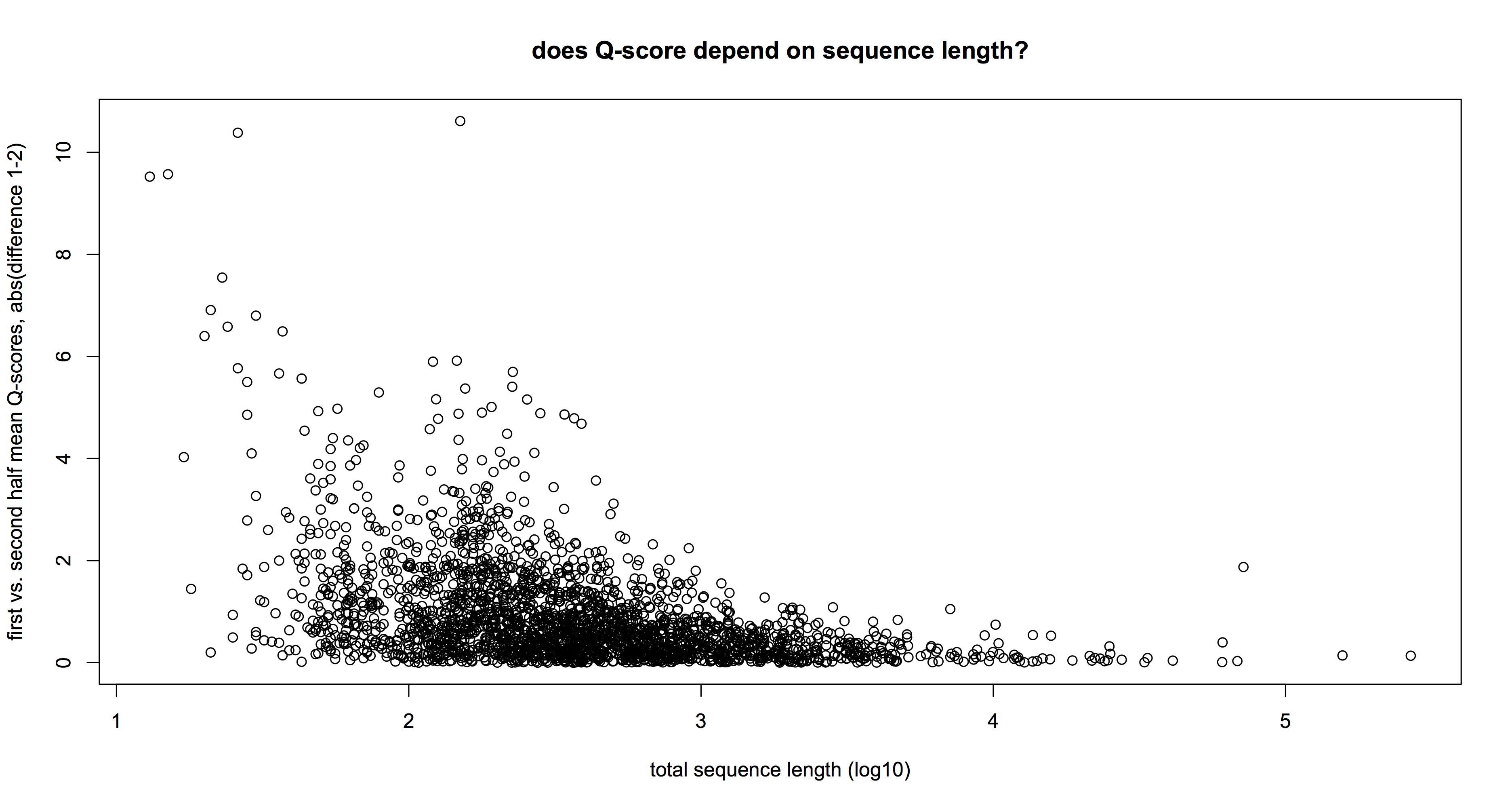
As for accuracy, well, the Q-scores are definitely lower in the 1D prep; around half the 2D as we expected. On the other hand, they were still matching reference databases via BLAST/BLAT/BWA quite happily – so if your application was ID, who cares? Equally, combining mega-long 1D reads with more shorter but accurate reads could be a good way to close gaps in a de novo genome sequencing project. One technical point – there is definitely a lot more relative variation in the Q-scores for short (<1000bp) reads than for longer ones: the plot above shows the absolute difference in mean Q-scores in (first – vs – second) halves of a subset of 2,300 1D reads. You can see that below 1kb, Q-score variation exceeds 4 (bad news when mean is about there) while longer reads have no such effect (quick T-test confirms this).
So in short, the 1D prep is great if you just want to get some DNA on your screen ASAP, and/or long reads to boot. In fact, if you came up with a way to size-select all the short gubbins out before sequencing, you’d have one mega-cool sequencing protocol! What about the last bit of our test – seeing if a quick and dirty extraction could work, too? The results were… mixed-to-poor. Gel electrophoresis and Qubit both suggested the extracted DNA was pretty poor quality/concentration, and if we didn’t believe them, the gloopy, aromatic, multicoloured liquid in the tubes supplied direct evidence to our eyes. So rather than test those samples first (and risk damaging a perfectly good flowcell early on in the experiment), we held them back until the end when only a handful of pores were left. In this condition it’s hard to say whether they worked, or not: the 50 or so reads we got over an hour or two from fewer than 10 pores is a decent haul, and some of them had some matches to congenerics in BLAST, but we didn’t really give them a full enough test to be sure.
Either way, we’ll be playing around with this more in the months to come, so watch. This. Space…
*Edit: Oxford Nanopore have recently announced that the rapid kit will be out in April for general purchase.
**Again working, for better or worse, with fellow bio-beardyman Dr. Alex Papadopulos. Hi Alex! This work funded by a Royal Botanic Gardens, Kew Pilot Study Grant.
Quick one this, as it’s a tricky problem I keep having to Google/SO. So I’m posting here for others but mainly myself too!
Here’s the situation: you have a folder (with, ooh, let’s say 140,000 separate MinION reads, for instance…) which contains a subset of files you want to move or copy somewhere else. Normally, you’d do something simple like a wildcarded ‘cp’ command, e.g.:
1 | host:joeparker$ cp dir/*files_I_want* some/other_dir |
Unfortunately, if the list of files matched by that wildcard is sufficiently long (more than a few thousand), you’ll get an error like this:
1 | -bash: /bin/cp: Argument list too long |
In other words, you’re going to have to be more clever. Using the GUI/Finder usually isn’t an option either at this point, as the directory size will likely defeat Finder, too. The solution is pretty simple but takes a bit of tweaking to work in OSX (full credit to posts here and here that got me started).
Basically, we’re going to use the ‘find’ command to locate the files we want, then pass each one in turn as an argument to ‘cp’ using ‘find -exec’. This is a bit slower overall than doing the equivalent as our original wildcarded command but since that won’t work we’ll have to lump it! The command* is:
1 | find dir -name *files_I_want* -maxdepth 1 -exec cp {} some/other_dir \;</p> |
Simple, eh? Enjoy 🙂
*NB, In this command:
I’ve recently been fiddling about and trying to fake an OSX-style GUI appearance in Linux Ubuntu MATE (15.04). This is partly because I prefer the OSX GUI (let’s be honest) and partly because most of my colleagues are also Mac users mainly (bioinformaticians…) and students in particular fear change! The Mac-style appearance seems to calm people down. A bit.
The specific OS I’m going for is 10.9 Mavericks, because it’s my current favourite and nice and clear. There are two main things to set up: the OS itself and the appearance. Let’s take them in turn.
I’ve picked Ubuntu (why on Earth wouldn’t you?!) and specifically the MATE distribution. This has a lot of nice features that make it fairly Mac-y, and in particular the windowing and package management seem smoother to me than the vanilla Ubuntu Unity system. Get it here: https://ubuntu-mate.org/vivid/.* The installation is pretty painless on Mac, PC or an existing Linux system. If in doubt you can use a USB as the boot volume without affecting existing files; with a large enough partition (the core OS is about 1GB) you can save settings – including the customisation we’re about to apply!
*We’re installing the 15.04 version, not the newest release, as 15.04 is an LTS (long-term stable) Ubuntu distribution. This means it’s supported officially for a good few years yet. [Edit: Arkash (see below) kindly pointed out that 14.04 is the most recent LTS, not 15.04. My only real reason for using 15.04 therefore is ‘I quite like it and most of the bugs have gone'(!)]
The MATE windowing system is very slick, but the green-ness is a bit, well, icky. We’re going to download a few appearance mods (themes, in Ubuntu parlance) which will improve things a bit. You’ll need to download these to your boot/install USB:
Now that we’ve got everything we need, let’s boot up the OS. Insert the USB stick into your Mac-envious victim of choice, power it up and enter the BIOS menu (F12 in most cases) before the existing OS loads. Select the USB drive as the boot volume and continue.
Once the Ubuntu MATE session loads, you’ll have the option of trialling the OS from the live USB, or permanently installing it to a hard drive. For this computer I won’t be installing to a hard drive (long story) but using the USB, so customising that. Pick either option, but beware that customisations to the live USB OS will be lost should you later choose to install to a hard drive.
When you’re logged in, it’s time to smarten this baby up! First we’ll play with the dock a bit. From the top menu bar, select “System > Preferences > MATE Tweak” to open the windowing management tool. In the ‘Interface’ menu, change Panel Layouts to ‘Eleven’ and Icon Size to ‘Small’. In the ‘Windows’ menu, we’ll change Buttons Layout to ‘Contemporary (Left)’. Close the MATE Tweak window to save. This is already looking more Mac-y, with a dock area at the bottom of the screen, although the colours and icons are off.
Now we’ll apply some theme magic to fix that. Select “System > Preferences > Look and Feel > Appearance”. Now we can customise the appearance. Firstly, we’ll load both the ‘Ultra-Flat Yosemite Light’ and ‘OSX-MATE’ themes, so they’re available to our hybrid theme. Click the ‘Install..’ icon at the bottom of the theme selector, you’ll be able to select and install the Ultra-Flat Yosemite Light theme we downloaded above. It should unpack from the .zip archive and appear in the themes panel. Installing the OXS-MATE theme is slightly trickier:
We’ll create a new custom theme with the best bits from both themes, so click ‘Custom’ theme, then ‘Customise..’ to make a new one. Before you go any further, save it under a new name! Now we’ll apply changes to this theme. There are five customisations we can apply: Controls, Colours, Window Border, Icons and Pointer:
Save the theme again, and we’re done! Exit Appearance Preferences.
Finally we’ll install Solarized as the default terminal (command-line interface) theme, because I like it. In the MATE Terminal, Unzip the solarized-mate-terminal archive, as sudo. Enter the directory and simply run (as sudo) the install script using bash:
$ sudo unzip solarized-mate-terminal
$ cd solarized-mate-terminal
$ bash solarized-mate.sh
Close and restart the terminal. Hey presto! You should now be able to see the light/dark Solarized themes available, under ‘Edit > Profiles’. You’ll want to set one as the default when opening a new terminal.
Later, I also installed Topmenu, a launchpad applet that gives an OSX-style top-anchored application menu to some linux programs. It’s a bit cranky and fiddly though, so you might want to give it a miss. But if you have time on your hands and really need that Cupertino flash, be my guest. I hope you’ve had a relatively easy install for the rest of this post, and if you’ve got any improvements, please let me know!
Happy Tweaking…
 Molecular phylogenetics – uncovering the history of evolution using signals in organisms’ genetic sequences – is a powerful science, the latest expression of the human desire to understand our common origins. But for all its achievements, I’d always felt something was, well lacking from my science. This week we’ve been experimenting with the MinION for the first time, and now I know what that is: immediacy. I’ll return to this theme later to explain how exciting this realisation is, but first we better ask:
Molecular phylogenetics – uncovering the history of evolution using signals in organisms’ genetic sequences – is a powerful science, the latest expression of the human desire to understand our common origins. But for all its achievements, I’d always felt something was, well lacking from my science. This week we’ve been experimenting with the MinION for the first time, and now I know what that is: immediacy. I’ll return to this theme later to explain how exciting this realisation is, but first we better ask:
What is a MinION?
NGS nerds know about this already, of course, but for the rest of us: The MinION (minh-aye-on) is a USB-connected device marketed by a UK company, Oxford Nanopore, as ‘a portable real-time biological analyser’. Yes – that does sound a lot like a tricorder, and for good reason: just like the fictional device, it promises to make the instant identification of biological samples a reality. It does this using a radically different new way to ‘read’ DNA sequences from a liquid into a letters (‘A, C, G, T‘) on a computer screen. Essentially, individual strands of DNA are pulled through a hole (a ‘nanopore’) in an artificial membrane, like the membranes that surround every living cell. When an electric field is applied to the membrane, the individual DNA letters (actually, ‘hexamers’ – 6-letter chunks) passing through the nanopore can be directly detected as fluctuations in the field, much like a magnetic C-60 cassette tape is read by a magnetic tape head. The really important thing is that, whereas other existing DNA-reading (‘sequencing’) machines take days-to-weeks to read a sample, the MinION produces output in minutes or even seconds. It’s also (as you can see in the picture, above) a small, wait tiny device.
Real-time
This combination of fast results and small size makes the tricorder dream possible, but this is more than just a gadget. Really, having access to biological sequences at our fingertips will completely change biology and society in many ways, some of which Yaniv Erlich explored in a recent paper. In particular, molecular evolutionary biologists will soon be able to interact with their subject matter in ways that other scientists have long taken for granted. What I mean by this is that in many other empirical disciplines, researchers (and schoolkids!) are able to directly observe, or easily measure, the phenomena they study. Paleontologists dig bones. Seismologists feel earthquakes. Zoologists track lions. And so on. But until now, the world of genomic data hasn’t been directly observable. In fact reading DNA sequences is a slow, expensive pain in the arse. And in such a heavily empirical subject, I can’t help but feel this is a hindrance – we are burdened with a galaxy of baroque models to explain variations in the small number of observations we’ve made, and I’ve got a hunch more data would actually consign many of them to the dustbin.
Imagine formulating, testing, and validating or discarding genetic hypotheses as seamlessly as an ecologist might survey a new forest…

Actual use
That’s the spiel, anyway. This week we actually used the MinION for the first time to sequence DNA (I’d been running some other technical tests for a couple of months, but this was our first attempt with real samples) and since it seems lots more colleagues want to ask about this device than have had a chance to use it, I thought I’d share our experiences. I say ‘we’ – work took place thanks to a Pilot Study Fund grant at the Royal Botanic Gardens, Kew, in collaboration with Dr. Alex Papadopulos (and really useful input from Drs. Andrew Helmstetter, Pepijn Kooij and Bryn Dentinger). It’s worth pointing out that although there’s a lot of hype about the device, it is still technically in a prototype/public-beta type phase, so things are changing all the time in terms of performance, etc.
 First up, the size. The pictures don’t really do it justice… compared with existing machines (fridge-sized, often) the MinION isn’t small, it’s tiny. The thing itself, plus box and cable, would easily fit into a side pocket on any laptop bag. So the ‘portable’ bit is certainly true, as far as the platform itself is concerned. But there’s a catch – to prepare a biological sample for sequencing on the MinION, you first have to go through a fairly complicated set of lab steps. Known collectively as ‘library preparation’ (a library here meaning a test tube containing a set of DNA molecules that have been specially treated to prepare them to be sequenced), the existing lab protocol took us several hours the first time. Partly that was due to the sheer number of curious onlookers, but partly because some of the requisite steps (bead cleanups etc) just need time and concentration to get right. None of the steps is particularly complicated (I’m crap at labwork and just about followed along) but there’s quite a few, and you have to follow the steps in the protocol carefully.
First up, the size. The pictures don’t really do it justice… compared with existing machines (fridge-sized, often) the MinION isn’t small, it’s tiny. The thing itself, plus box and cable, would easily fit into a side pocket on any laptop bag. So the ‘portable’ bit is certainly true, as far as the platform itself is concerned. But there’s a catch – to prepare a biological sample for sequencing on the MinION, you first have to go through a fairly complicated set of lab steps. Known collectively as ‘library preparation’ (a library here meaning a test tube containing a set of DNA molecules that have been specially treated to prepare them to be sequenced), the existing lab protocol took us several hours the first time. Partly that was due to the sheer number of curious onlookers, but partly because some of the requisite steps (bead cleanups etc) just need time and concentration to get right. None of the steps is particularly complicated (I’m crap at labwork and just about followed along) but there’s quite a few, and you have to follow the steps in the protocol carefully.
Performance
So how did the MinION do? We prepared two libraries; one from a control sample of bacteriophage lambda (a viral genome, used to check the lab steps are working) and another from Silene latifolia, a small flowering plant and one of Alex’s faves. The results were exciting. In fact they were nerve-wracking – initially we mixed up the two samples by mistake (incredible – what pros…) and were really worried when, after a few hours’ sequencing, not a single DNA read matching the lambda genome had appeared. After a lot of worrying, and restarting various analyses, including the MinION itself, we eventually realised our mistake, reloaded the MinION with the other sample and – hey presto! – lambda reads started to pour out of the software. In the end, we were able to get a 500x coverage really quickly (see reads mapped to the reference genome, below):
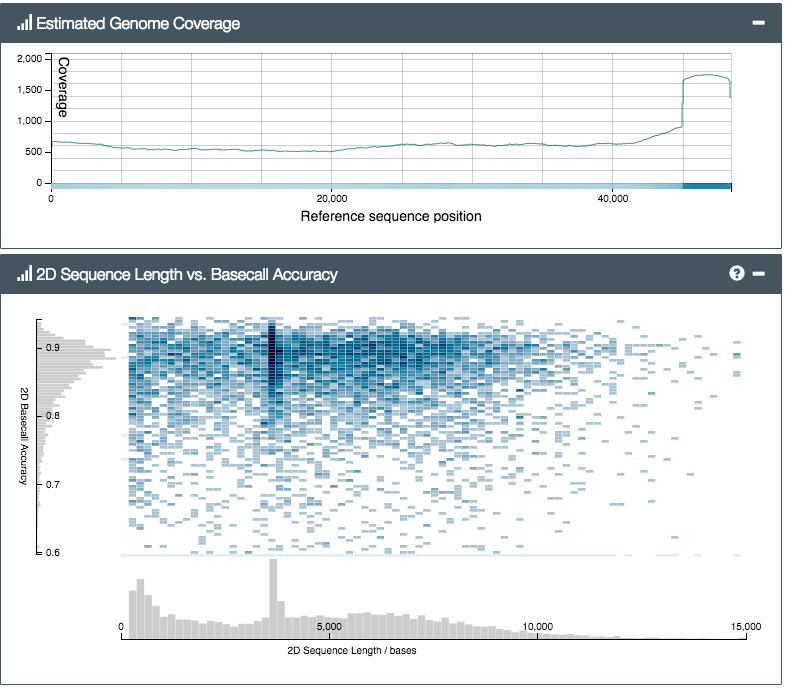
You can see from the top plot that we got good even coverage across the genome, while the bottom plot shows an even (ish) read length distribution, peaking around 4kb. We’d tried for a target size of 6kb (shearing using g-TUBEs), so it seems the actual output read size distribution is lower than the shearing target – you’d need to aim higher to compensate. Still, we got plenty of reads longer than 20, or even 30,000 base-pairs (bp) – from a 47kbp genome this is great, and much much much longer than typical Illumina paired-end reads of ~hundreds of bp.
Later on the next day, we decided we’d done enough to be sure the library preparation and sequencing were working well, so we switched from the lambda control to our experimental (S.latifolia) sample. Again, we got a good steady stream of reads, and some were really long, over 65,000bp. Crucially, we were able to BLAST these against NCBI and get hits against the NCBI public sequence database and get Silene hits straight back. We were also able to map them onto the Silene genome directly using BWA, even with no trimming or masking low-quality regions. Overall, we got nearly a full week’s sequencing from our flowcell, with ~24,000 reads at a mean length of ~4kb. Not bad, and we’d have got more if we hadn’t wasted the best part of the run sodding about while we troubleshot our library mix-up at the start (the number of active pores, and hence sequencing throughput, declines over time).
The bad: There is a price for these long, rapid reads. Firstly, the accuracy is definitely lower than Illumina – although the average lambda library sequencing accuracy in our first library was nearly 90%, on some of our mapped Silene reads it dropped much lower – 70% or so. This has been well documented, of course. Secondly, the *big* difference between the MinION in use and a MiSeq or HiSeq is the sheer level of involvement needed to get the best from the device – whereas both Illumina machines are essentially push-button operated, the MinION seems to respond well if you treat it kindly (reloading etc) and not if you don’t (introducing air bubbles while loading seemed to wreck some pores)
The very, very, good: Ultimately though, we came away convinced that these issues won’t matter at all in the long run. ONT have said that the library prep and accuracy are both boing improved, as are flowcell quality (prototype, remember). Most importantly the MinION isn’t really a direct competitor to the HiSeq. It’s a completely different instrument. Yes, they both read DNA sequences, but that’s where the similarity ends. The MinION is just so flexible, there’s almost an infinite variety of uses.
In particular, we were really struck that the length of the reads means simple algorithms like BLAST can get a really good match from any sample with just a few hundreds, or tens, of reads from a sample. You just don’t need millions of 150bp reads to match an unknown sample to a database with reads this long! Coupled with the fact that the software is real-time and flowcells can be stopped and started, and you have the bones of a really capable genetic identification system for all sorts of uses; disease outbreaks (in fact see this great work on Ebola using MinION); customs control of endangered species; agriculture; brewing – virtually anything, in fact, where finding out about living organisms’ identity or function is needed.
The future.
There’s absolutely no question at all: really, these devices are the future of biology. Maybe the MinION will take off (right now, the buzz couldn’t be hotter), or a competitor will find a way to do even more amazing things. It doesn’t matter how it comes about, though – the next generation of biologists really will be living in a world where, in 10, 15, 20 years, at most, the sheer ubiquity of sequencers like this will mean that most, if not all eukaryotes’ genomes will have been sequenced, and so can be easily matched against an unknown sample. If that sounds fantastic, consider that the cumulative sequencing output of the entire world in 1988 amounted to a little over 100,000 sequences, a yield equivalent to a single good MinION run. And while most people in 1988 thought that, since the human genome might take 30 or 40 years to sequence, there was little point in even starting, a few others looked around at the new technology, its potential, and drew a different conclusion. The rest is history…
General science talk about the potential of real-time phylogenomics, delivered at the Jodrell Lecture Theatre, Kew Gardens, November 2nd 2015
Slides [SlideShare]: cc-by-nc-nd
[slideshare id=54651010&doc=real-time-phylogenomics-joeparker-151102162613-lva1-app6892]
Had a heated discussion with a friend the other day. I went to a school, where ‘exam techniques’ were part of the standard toolkit given to students to get them the best possible grades at GCSE, A-levels, and beyond. She didn’t, and so hadn’t ever heard of a special ‘technique’ for exams until uni. She felt robbed – why should one group of students get an advantage over the others, because their school taught them how to cheat the system?
Well, it’s a fair point; but my reply was that exam techniques really aren’t that complicated. In fact, you can boil most of them down to three simple rules: Answer the Question, Plan Your Time and Plan Your Answers. Look, I can even explain each one in 100 words or fewer 😉
Every stack of exam scripts that’s ever been marked from Socrates* onwards contains at least one howling stink-bomb of a perfect answer. The student has a deep and broad understanding of their subject. The answer is comprehensive, incisive, and backed up by copious references. Unfortunately, because they’ve misread the question and gone off on a tangent, you sigh, marvelling at the intelligence that manages to completely grasp a difficult concept like mitochondrial introgression, but utterly failed to comprehend the text of the exam paper. Nil marks. Don’t let it be you: read the question carefully and give the examiner only what they’ve asked for.
*Socrates may not have actually sat a single GCSE, but you get the point.
This one is too obvious for words, but you must practice and be self-disciplined in the exam for it to work. Basically, most students subconsciously assume that the relationship between time spent on a question and marks collected is linear, something like this:
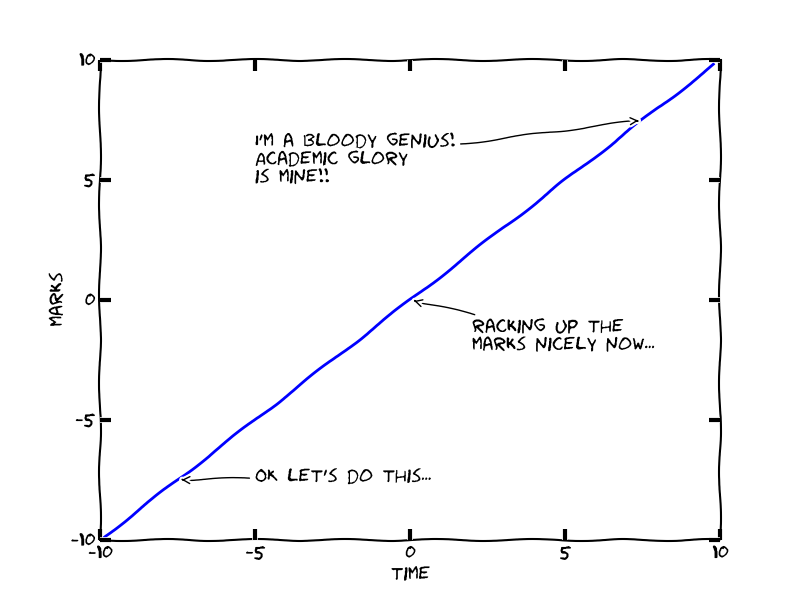
(Edit: err, ignore the axes’ values… oops!)
Wrong. It’s minimally true that the longer you spend on a question, the more marks you’ll probably* get. But most exams have more than one question, so you must balance trying to get top marks on, say, Question 1, with getting at least some marks on Questions 2, 3 & 4! This is why it’s important to get a grip on the real relationship between effort and marks. It looks a lot more like this:
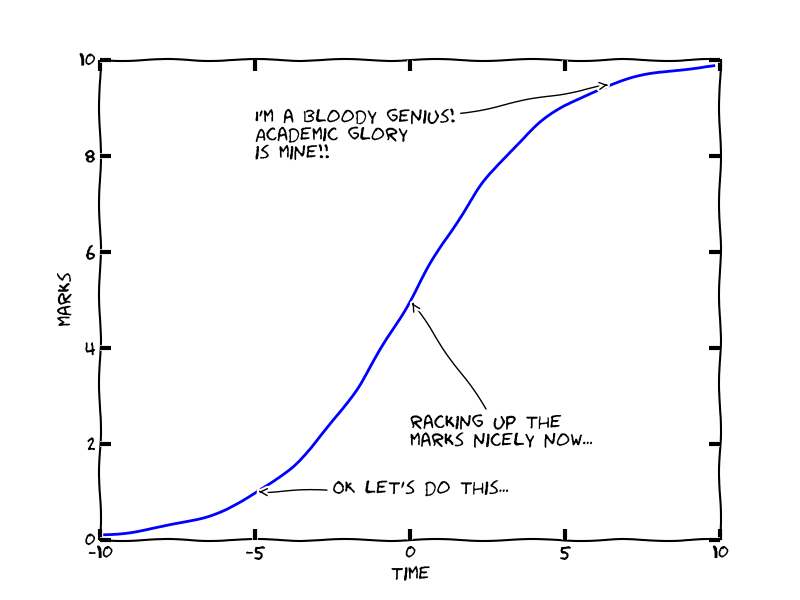
Can you see what’s going on here? Most exam questions are deliberately structured so that getting a third-class (‘D’) grade is relatively easy, then marks are awarded more-or-less linearly up to the top of an upper-second (‘B). Finally, first-class (‘A’) grade answers usually require substantially more insight – and deliberately and rightly so, since these grades are supposed to mark out the very best exam scripts, usually around 5-10% of the class at most.
In other words, if you spend half of a 100-minute, four-question exam on Question 1, you might get an A for maybe 25% of the marks… but at the cost of achieving a C, maximum, on the other three questions. You’ll be lucky to average a C. Plan your time.
*’probably’ because after you’ve spent too long on a question there’s a temptation to start chucking in the kitchen sink as well, and after a while you’re at risk of saying something stupid or wrong which might actually cost you marks.
When you turn over the exam paper and pick up your pen, your head is likely buzzing – with caffeine, formulae and references you’ve crammed in at the last minute, and possibly with the dull ache of worry that your Mum will kill you if you don’t pass this exam. You want to calmly and methodically, putting the first two exam techniques to devastating effect and dazzling your examiners. You’ll actually probably grab the first question you like the look of and start writing immediately.
This is almost guaranteed to leave you sweating in a heap of confusion half-way through the exam, when you look up from polishing your first answer, realise time’s running out, and start flailing through the others. Without an answer plan you’ll find it hard to stick to time, and in the case of longer questions you’ll be more likely to stray off-piste as well.
Instead, invest some of the exam time into making a plan – with bullet points – for each answer. I always spend at least 5% of the time for each answer writing a plan. Do this on your answer script too, so that in the nightmare scenario that you run out of time the examiner can at least give you some marks. For instance:
Q2: Explain what is meant by Muller’s Ratchet, in the context of natural selection (30% of marks)
A: Outline answer:
- Muller’s Ratchet (Muller, 1964) is accumulation of deleterious mutations in asexual populations
- ‘Deleterious’ means lowering fitness of an organism
- Asexual populations (or chromosomes) do not exchange genetic material in meiosis by recombination, unlike sexually-reproducing populations/chromosomes.
- Mutations in genetic material occur over time at random
- Most changes have little effect, or slightly deleterious
- Some highly deleterious or advantageous
- Natural selection filters randomly-occurring mutations. Under ‘neutral’ selection the effect of all mutations is negligible. Under ‘positive selection’ advantageous mutations’ benefit leads to their higher relative occurrence through evolutionary time. Under ‘negative selection’ deleterious mutations penalised highly
- Muller’s Ratchet therefore implies that asexually-reproducing populations subject to negative selection will be disadvantaged compared to sexually-reproducing populations, as they cannot filter out deleterious mutations through recombination.
It probably took about 5 minutes to think about and write that plan – but it would likely get at least half-marks, on its own…
In many cases you will be able to get an idea of how many questions, and which topics, the exam will contain (using nefarious tricks like simply asking the course convenor). Armed with this, you can plan your overall exam strategy, with times. Something like this:
Plan for a 2 hour exam with 3 questions (choice of 7) starting at 10:30am:
You can take your exam plan in with you (at least in your head) so you don’t waste valuable time trying to work out your timings in the real thing. You’ll also feel more confident and in control of your performance.
The above three techniques will help you make the most of your hard-won knowledge (you did revise the content too, right?) but – trust me on this one, cos I’m an exam machine – you’ll be utterly unable to put them to effect without exam practice. This is probably the biggest difference between schools which actually spend precious teaching time on exam practice, and those which simply point students to blogs like this one as part of their revision.
Get a group of mates on the same course together, get a stack of past exam scripts, and practice in exam conditions. Compare, and mark each others’ scripts. Then repeat, again and again.
Last of all, don’t overdo the coffee. Good luck!
I’ve just seen the proposal from Westminster Council on a section of the Quietway Grid in their patch from Covent Garden to Waterloo Bridge. A patch, lest we forget that’s used by a lot of bikes:

I’m really pushed for time this week so I won’t write a detailed post on this. In short it is terrible. Virtually nothing proposed at all, and what little there is is for pedestrians, not cyclists! Loads of car parking retained, no modal filters.
Worst of all, on Waterloo Bridge and the Strand junction itself there is nothing, absolutely nothing to protect cyclists. A line of paint on the road is all that’s going to protect you from busses, lorries, coaches and cars. This is 2015, not 1995. The council are mad.
This is the absolute centre of civic life in London and if there isn’t a case for pedestrianising large swathes of it (allowing black cabs to some bits, and perhaps loading/deliveries outside peak hours), then I can’t think where in the UK it would be appropriate to pedestrianise.
As I say, I’m in a mega hurry, so here’s my pasted consultation response:
What on earth is the Council doing? These are appalling proposals. The Council seem to have utterly disregarded the LCDS2 and LCC recommendations for Quietway provision and instead decided to do virtually nothing. Where money *is* spent it is on pavement widening to benefit pedestrians, not cycling. There is no reason whatsoever why these streets, right in the heart of London, should be accessed by private cars. Taxis, yes, deliveries/loading yes (at prescribed times) but all private cars, all the time? Madness.
Unless additional permeability measures benefit bikes, and modal filters discourage cars, they will struggle to create a cycle-friendly network that encourages cycling as an everyday activity.
Furthermore lots of car parking and loading bays *are* retained which (contrary to stated on the plan) create pinch points and encourage car use. Instead the spaces should be inset to the pavement if they are to be retained at all.
Section 1 (Bow St etc) – there is hardly anything here proposed to comment on. No modal filters, nothing. Strongly oppose.
Section 2 (Covent Garden etc) – again nothing for cycling to comment on at all. The gain of raised tables will be more than offset by retained car parking and to include footway resurfacing as part of a ‘cycling scheme’ is laughable.
Section 3 (Strand) – the only element of this proposal that is genuinely welcome is dropping the junction across the Strand to carriageway level. The rest of the section propsal is nonsense (again, why are private cars to be allowed to drive and park outside the Lyceum, exactly?!) and the unprotected central feeder lane on Waterloo Bridge n/bound is utter madness. It belongs in the 1990s and will be incredibly dangerous – encouraging some novices to the centre of the traffic whilst providing zero protection. If a traffic lane can be lost then why not provide a fully segregated facility on this incredibly busy bridge?
Section 4 (Waterloo bridge northern end) – this proposal is so close to criminally liable I’m amazed the Council even let it out into the public domain. Given the documented incredibly high cycle mode share on this bridge, to protect cycle traffic with a **single white line of paint on the road** eg a mandatory on-road cycle lane, when there is a central reservation present, is beyond belief. This won’t do anything, *at* *all* to improve safety.
TO call this a cycling scheme is wholly disingenuous and the Council should consider seriously whether they have opened themselves to a judicial review by doing so.
Southwark Cyclists reject these proposals.
Southwark Council’s proposals for Camberwell Green alterations do nothing to address serious and worsening safety issues for cyclists.
These largely cosmetic alterations also miss several opportunities to substantially improve crossing times and safety for pedestrians.
These proposals assume increasing traffic flow when TfL’s own figures show car ownership, use, and miles travelled are all decreasing in this part of London. The Council’s own figures and transport policy forecast a rise in cycle numbers.
We urge Southwark Council and Transport for London to reconsider their approach to this key junction and the Camberwell Green area. Space for cycling at the junction and/or a cycle bypass are feasible alternatives to their proposals.
Sensible Alternatives
A number of sensible alternatives exist which could be built in a similar amount of time and disruption, and would greatly improve the safety and convenience of the area for cyclists and pedestrians alike:

The public environment. Firstly, even the council recognise that the Green itself is a retail and services destination. Thousands of local people use the Green every day for shopping, the library, pharmacy, clinic and courts. But the public environment on Camberwell Church Street and Denmark Hill (outside Butterfly Walk) is intimidating for pedestrians and cyclists alike, with trucks and coaches hurtling down six lanes of traffic at 30mph+, as this picture shows. It’s hard to cross and no wonder so many people prefer to drive short distances to these shops. The proposal does nothing to improve the environment – although the pavement itself will get some expensive new stone, nothing will be done to make it easier or more inviting to get to the Green by bike or on foot, or cross the road once you’re there. Southwark Cyclists propose the town centre, one of the historic local greens of South London, be more imaginatively redesigned as a retail and services destination where pedestrians and cyclists come first. This can be done while maintaining traffic capacity, but calming it.

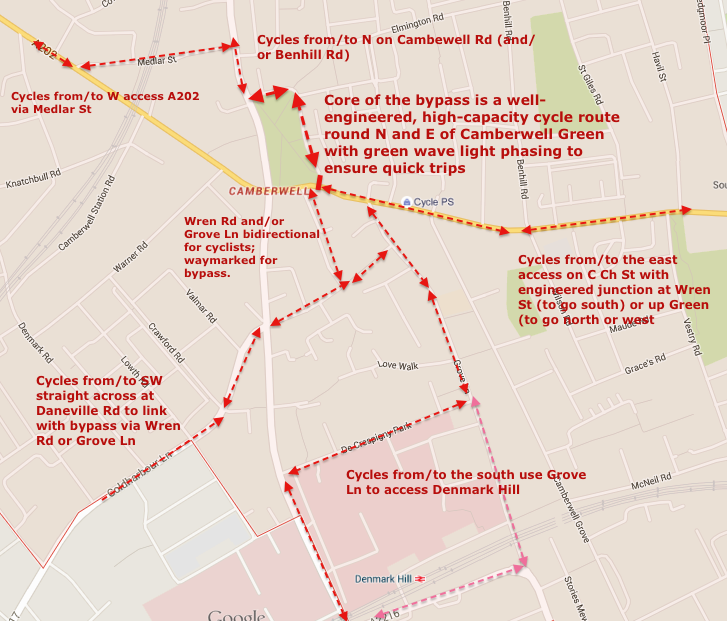
A bus hub – in Orpheus Street, not the high street. One of the council’s motivations for doing anything at all is the large numbers of pedestrians waiting for busses on the pavement outside the shopping centre. Often these spill out into the road; it’s unsafe and hard to get past with a pushchair or wheelchair. Instead of the council’s plans (which actually add hardly any space at all on the pavement in question, and do nothing to calm the traffic at all) we suggest several bus stops could be moved 10-20m down, into Orpheus Street, creating a local bus hub. With proper lighting and other features, this could also be a much safer place to wait, and would create additional retail or services frontages in Orpheus Street, which at the moment is a barren alley with just the art shop. Why move the bus stops at all? Well, moving them from the high street (Denmark Hill) would make the crossing simpler for pedestrians, make driving easier for cars (no busses cutting in and out suddenly), make cycling safer (by freeing space for a bike lane) and the environment far more pleasant for everyone.
Basically, there are several options, far better than the Council’s plans. Reject these plans today (Thurs 13th August closing date) and send them back to the drawing board!
Presentation on lightweight bioinformatics (Raspi / cloud computing) for real-time field-based analyses.
Presented at iEOS2015, St. Andrews, 3-6th July 2015.
Slides [SlideShare]: cc-by-nc-nd
[slideshare id=50251856&doc=joeparkerlightweightbioinformatics-150707112254-lva1-app6892]

{kind=link}