
My last MinION post described our first experiments with this really cool new technology. I mentioned then that their standard library prep was fairly involved, and we heard that the manufacturers, Oxford Nanopore, were working on a faster, simpler library prep. We got in touch and managed to get an early prototype of this kit for developers*, so we thought we’d try it out. So our** experiments had three aims:
- Try out this new rapid kit
- Try out different extraction methods, to see how they worked with the kit
- See if we could sequence some fairly damaged DNA with the kit
This is a lot of combinations to perform over a few days on one sequencing platform! Our experience from the last run gave us hope we could manage it all (we did, but with a lot of headaches over disk space – it turns out one drawback of being able to run multiple concurrent sequencing runs is hard-drive meltdown, unless you’re organised from the start – oops). In fact, to keep track of all the reads we added an ‘index’ function to the Poretools package. I really recommend you use this if you’re planning your own work.
We had eight samples to sequence, a 15-year-old dried fungarium specimen of Armillaria ostoye (likely to be poor quality DNA; extracted by Bryn Dentinger with a custom technique); some fresh Silene latifolia (Qiagen-extracted, which we’d used successfully with the previous, ‘2D’ library prep); and six arbitarily-selected plant samples, both monocots and dicots, extracted by boiling with Chelex beads (more about them at the end).

First we prepared normal ‘2D’ libraries from the Silene and Armillaria. These performed as expected from our December experiments, even the Armillaria giving decent numbers and lengths of reads (though not as many as we hoped, with some indication of worse Q-scores. We put this down to nicks in the fungarium DNA, and moved on to the 1D preps while the sequencer was still running.
The ‘1D’ in the rapid kit (vs ‘2D’ in the normal kit) refers to how many DNA strands are sequenced; in the 2D version, both forward and reverse-complement strands are sequenced. This is slower to prepare (extra adapters etc to link the two strands for sequencing) and also runs through the MinION more slowly (twice as much DNA, plus a hairpin moment) but is roughly twice as accurate, since each base is read twice. The 1D kit, on the other hand, results in single-stranded fragments, meaning we could expect lower accuracy traded off for higher speed. And the rapid kit really was fast – starting from purified extracted DNA, we added all the necessary adapters for sequencing in well under 15mins, ready to sequence.
The sequencing itself (for both the Armillaria and the Silene) went spectacularly well. Remember, this is unsheared genomic DNA, and imagine our surprise when we started to see 25, then 50, then 100, then 150kb reads come off the sequencer – many mapping straight away to reference genomes! It turns out that the size distribution of the 1D prep is much more long-tailed than the 2D/g-TUBE one. In fact, whereas the the 2D library looks like a normal-ish gamma, the 1D reads are more like an inverse exponential – lots of short stuff and then a very long tail with some mega-whopper reads in. Reads so long, in fact, that mapping them the same way as Illumina short-read data would be a bit bonkers…
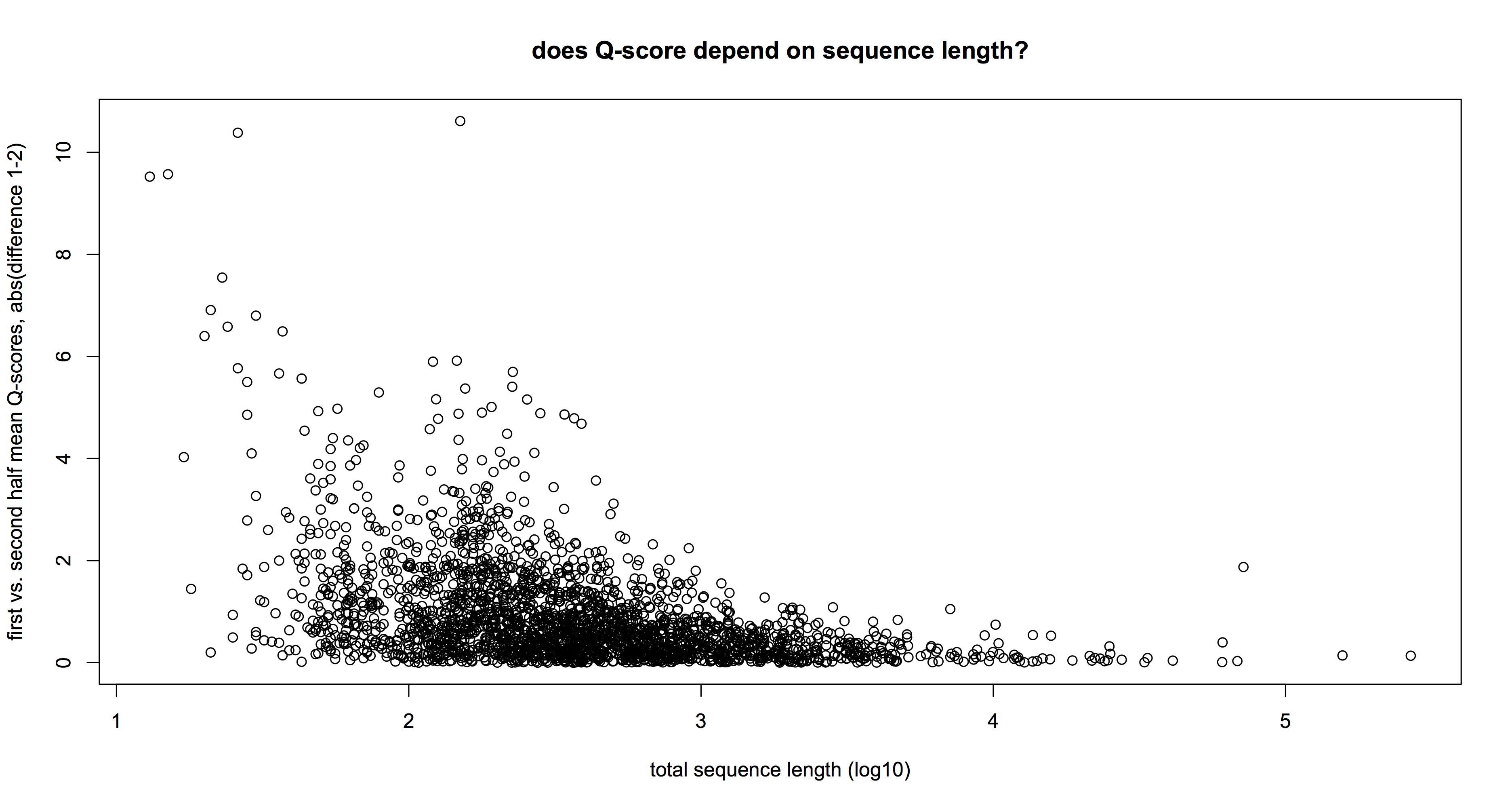
As for accuracy, well, the Q-scores are definitely lower in the 1D prep; around half the 2D as we expected. On the other hand, they were still matching reference databases via BLAST/BLAT/BWA quite happily – so if your application was ID, who cares? Equally, combining mega-long 1D reads with more shorter but accurate reads could be a good way to close gaps in a de novo genome sequencing project. One technical point – there is definitely a lot more relative variation in the Q-scores for short (<1000bp) reads than for longer ones: the plot above shows the absolute difference in mean Q-scores in (first – vs – second) halves of a subset of 2,300 1D reads. You can see that below 1kb, Q-score variation exceeds 4 (bad news when mean is about there) while longer reads have no such effect (quick T-test confirms this).
{kind=link}
{kind=link}
{kind=link}
So in short, the 1D prep is great if you just want to get some DNA on your screen ASAP, and/or long reads to boot. In fact, if you came up with a way to size-select all the short gubbins out before sequencing, you’d have one mega-cool sequencing protocol! What about the last bit of our test – seeing if a quick and dirty extraction could work, too? The results were… mixed-to-poor. Gel electrophoresis and Qubit both suggested the extracted DNA was pretty poor quality/concentration, and if we didn’t believe them, the gloopy, aromatic, multicoloured liquid in the tubes supplied direct evidence to our eyes. So rather than test those samples first (and risk damaging a perfectly good flowcell early on in the experiment), we held them back until the end when only a handful of pores were left. In this condition it’s hard to say whether they worked, or not: the 50 or so reads we got over an hour or two from fewer than 10 pores is a decent haul, and some of them had some matches to congenerics in BLAST, but we didn’t really give them a full enough test to be sure.
Either way, we’ll be playing around with this more in the months to come, so watch. This. Space…
*Edit: Oxford Nanopore have recently announced that the rapid kit will be out in April for general purchase.
**Again working, for better or worse, with fellow bio-beardyman Dr. Alex Papadopulos. Hi Alex! This work funded by a Royal Botanic Gardens, Kew Pilot Study Grant.